Intermediate Tutorial - Prepare Data & Multi-Algorithm Runs
This tutorial builds on the introduction to SPRAS from the previous tutorial.
It guides participants through how to convert data into a format usable by pathway reconstruction algorithms, run multiple algorithms within a single workflow, and apply new tools to interpret and compare the resulting pathways.
You will learn how to:
Prepare and format data for use with SPRAS
Configure and run additional pathway reconstruction algorithms on a dataset
Enable post-analysis steps to generate post analysis information
Step 1: Transforming high throughput experimental data into SPRAS compatible input data
1.1 Example of high-throughput omic data
High-throughput omics technologies measure thousands of biological molecules in a single experiment, producing genome-, transcriptome-, or proteome-wide snapshots of cellular state. These measurements quantify how molecular abundance or activity changes across conditions or time points, generating large-scale datasets that can be used as input for pathway reconstruction.
An example dataset is EGF response mass spectrometry data [4], a proteomics dataset that measures peptide abundance after cells are stimulated with epidermal growth factor (EGF).
The experiment for this data was repeated three times, known as biological replicates, to ensure the results are consistent. Each replicate measures the abundance of peptides at different time points (0-128 minutes) to capture how protein activity changes over time.
Note
Mass spectrometry is a technique used to measure and identify proteins in a sample. It works by breaking proteins into smaller pieces called peptides and measuring their mass-to-charge ratio, which enables identifying which peptide is being measured. The data show how much of each peptide is present, which can show how protein phosphorylation abundances change under different conditions.
Since proteins interact with each other in biological pathways, changes in their phosphorylation abundances can reveal which parts of a pathway are active or affected.
Example of one peptide’s measurements in one of the biological replicates:
peptide |
protein |
gene.name |
modified.sites |
0 min |
2 min |
4 min |
8 min |
16 min |
32 min |
64 min |
128 mn |
|---|---|---|---|---|---|---|---|---|---|---|---|
K.n[305.21]AFWMAIGGDRDEIEGLS[167.00]S[167.00]DEEH.- |
Q6PD74,B4DG44,Q5JPJ4,Q6AWA0 |
AAGAB |
S310,S311 |
14.97 |
14.81 |
13.99 |
13.98 |
12.87 |
13.88 |
13.91 |
15.60 |
Omics data can serve as input for pathway reconstruction, but it must first be reformatted to match the input format and requirements of each algorithm.
1.2 What is the standardized input data?
A pathway reconstruction algorithm at minimum requires a set of input nodes (node_files) and an interactome (edge_files); however, each algorithm expects its inputs to follow a unique format.
Note
Input nodes are a set of molecules of interest, typically derived from high-throughput omics data.
An interactome is a network of known molecule-to-molecule interactions, typically compiled by aggregating experimental and curated data from public databases. It defines the set of possible edges that algorithms can draw on when reconstructing.
To simplify this process, SPRAS requires all input data in a dataset to be formatted once into a standardized SPRAS format. SPRAS then automatically generates algorithm-specific input files when an algorithm is enabled in the configuration file.
Note
Each algorithm uses the input nodes to guide or constrain the optimization process used to construct reconstruct subnetworks.
An algorithm maps these input nodes onto the interactome and identifies connecting paths between the input nodes to form subnetworks.
Pathway reconstruction algorithms differ in the inputs nodes they require and how they interpret those nodes to identify subnetworks.
Some use source and target nodes to defined start and end points.
Some use prizes, which assign numerical scores assigned to nodes of interest.
Some rely on active nodes, representing nodes that are significantly “on” under specific conditions.
An example of a node file required by SPRAS follows a tab-separated format:
NODEID prize sources targets active
A 1.0 True True
B 3.3 True True
C 2.5 True True
D 1.9 True True
Note
If a user provides only one type of input node but wants to run algorithms that require a different type, SPRAS can automatically convert the inputs into the compatible format:
Source-target nodes can be used with all algorithms by making a prize column set to 1 and an active column set to True.
Prize data can be adapted for active based algorithms by automatically making an active column set to True.
Active data can be adapted for prize based algorithms by making a prize column set to 1.
Along with differences in their inputs nodes, pathway reconstruction algorithms also interpret the input interactome differently.
Some algorithms can handle only fully directed interactomes. These interactomes include edges with a specific direction (A -> B).
Others work only with fully undirected interactomes. These interactomes have edges without direction (A - B).
And some support mixed-directionaltiy interactomes. These interactomes contain both directed and undirected edges.
Note
Directionality describes whether an edge in the interactome captures the direction of a biological interaction.
A directed edge (A -> B) means that molecule A acts on molecule B, but not the reverse, for example, a kinase phosphorylating its substrate or a transcription factor regulating a target gene.
An undirected edge (A - B) means that A and B interact, but the data does not specify which one acts on the other, for example, two proteins that bind each other in a complex.
SPRAS automatically converts the user-provided edge file (interactome) into the format expected by each algorithm, ensuring that the directionality of the interactome matches the algorithm’s requirements.
An example of an edge file required by SPRAS follows a tab-separated
format. where U indicates an undirected edge and D indicates a
directed edge:
A B 0.98 U
B C 0.77 D
Note
SPRAS supports multiple standardized input formats. More information
about input data formats can be found in the inputs/README.md
file within the SPRAS repository.
1.3 Preprocessing the omic data
Before analysis, we filter out peptides that are not present in all three replicates to ensure consistency across measurements. We then normalize each replicate so that intensity values are comparable and not biased by replicate-specific effects.
peptide |
protein |
gene.name |
modified.sites |
0 min |
2 min |
4 min |
8 min |
16 min |
32 min |
64 min |
128 mn |
replicate |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
K.n[305.21]AFWMAIGGDRDEIEGLS[167.00]S[167.00]DEEH.- |
Q6PD74,B4DG44,Q5JPJ4,Q6AWA0 |
AAGAB |
S310,S311 |
2.17 |
2.09 |
1.98 |
1.78 |
1.99 |
2.12 |
2.25 |
1.46 |
C |
K.n[305.21]AFWMAIGGDRDEIEGLS[167.00]S[167.00]DEEH.- |
Q6PD74,B4DG44,Q5JPJ4,Q6AWA0 |
AAGAB |
S310,S311 |
4.03 |
3.73 |
3.32 |
3.36 |
3.35 |
3.37 |
3.35 |
3.86 |
B |
K.n[305.21]AFWMAIGGDRDEIEGLS[167.00]S[167.00]DEEH.- |
Q6PD74,B4DG44,Q5JPJ4,Q6AWA0 |
AAGAB |
S310,S311 |
5.60 |
4.75 |
4.69 |
4.59 |
4.32 |
4.90 |
4.90 |
5.48 |
A |
1.4 Computing prizes
We can transform these measurements into prizes for pathway reconstruction. One approach is to calculate a p-value per peptide, which quantifies how likely changes in abundance happen by chance.
We use Tukey’s Honest Significant Difference (HSD) test to compare all time points and correct for multiple testing to get a p-value for every pair of time points.
peptide |
protein |
2min vs 0min |
4min vs 0min |
8min vs 0min |
16min vs 0min |
32min.vs.0min |
64min.vs.0min |
128min.vs.0min |
4min.vs.2min |
8min.vs.2min |
16min.vs.2min |
32min.vs.2min |
64min.vs.2min |
128min.vs.2min |
8min.vs.4min |
16min.vs.4min |
32min.vs.4min |
64min.vs.4min |
128min.vs.4min |
16min.vs.8min |
32min.vs.8min |
64min.vs.8min |
128min.vs.8min |
32min.vs.16min |
64min.vs.16min |
128min.vs.16min |
64min.vs.32min |
128min.vs.32min |
128min.vs.64min |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
K.n[305.21]ADVLEAHEAEAEEPEAGK[432.30]S[167.00]EAEDDEDEVDDLPSSR.R |
QQ6PD74,B4DG44,Q5JPJ4,Q6AWA0 |
0.67 |
0.25 |
0.14 |
0.12 |
0.52 |
0.76 |
0.84 |
0.99 |
0.93 |
0.90 |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
1.00 |
0.97 |
0.94 |
1.00 |
0.98 |
0.87 |
0.80 |
0.96 |
0.83 |
0.75 |
1.00 |
1.00 |
1.00 |
Peptides with lower p-values are more statistically significant and may represent biologically meaningful changes in phosphorylation over time.
To use these p-values as input node prizes, we transform them with
-log10(p-value) so that smaller p-values produce larger prize
scores.
Two adjustments are needed before the prizes are usable:
Collapsing temporal information: The dataset contains temporal measurements, but SPRAS does not include algorithms that use temporal information. For each peptide, we select the smallest p-value across all baseline-vs-time and consecutive time-point comparisons, since the smallest p-value represents the most significant change.
Resolving peptide-to-protein duplicates: A single protein can map to multiple peptides. For each protein, we assign the maximum prize value across all of its peptides.
Note
All node identifiers must use the same namespace across every part of a dataset.
For this dataset, all protein identifiers are converted to UniProt Entry Names, and the same conversion is applied to the interactome.
peptide |
protein |
uniprot entry name |
min p-value |
-log10(min p-value) |
|---|---|---|---|---|
K.n[305.21]AFWMAIGGDRDEIEGLS[167.00]S[167.00]DEEH.- |
Q6PD74,B4DG44,Q5JPJ4,Q6AWA0 |
AAGAB_HUMAN |
0.12392034609392 |
0.906857382317364 |
- Input node data put into a SPRAS-standardized format (and IDs mapped to UniProt
Entry Names):
NODE_ID prize
AAGAB_HUMAN 0.906857382
1.6 From prizes to sources, targets and actives
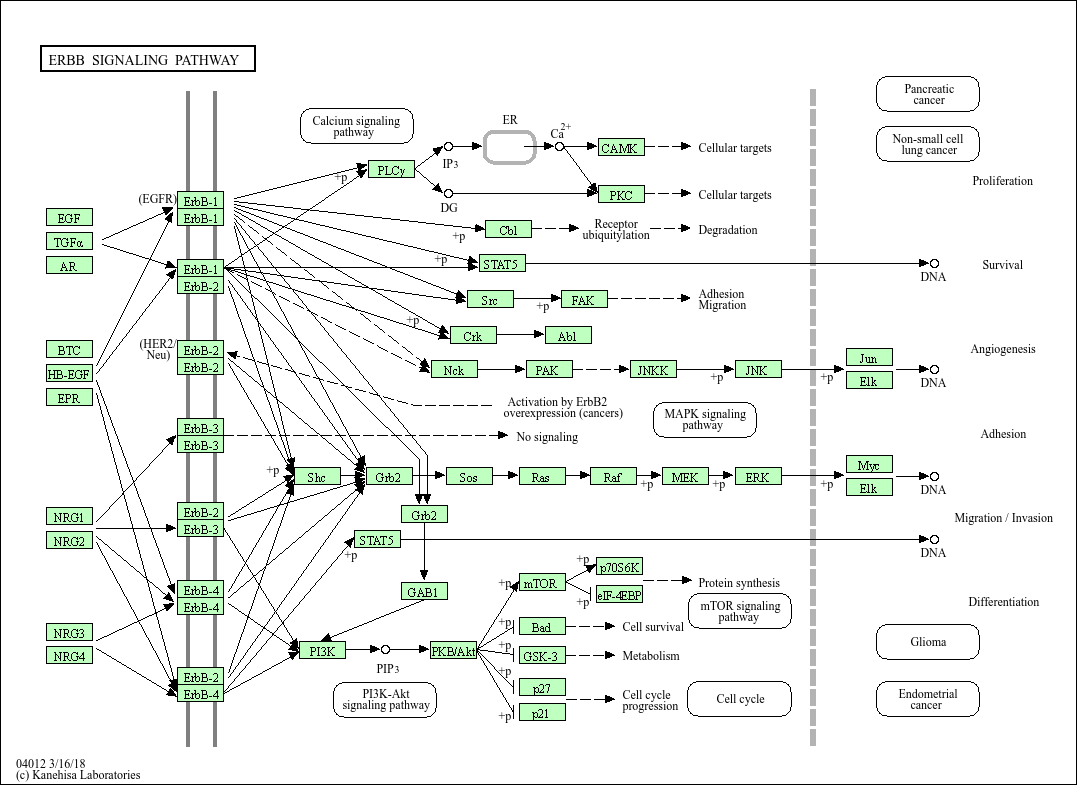
Using known pathway knowledge [1] [2] [3]:
EGF serves as a source for the pathway and was the experimental treatment.
EGF is known to initiate signaling, so it can be added and assigned a high score (greater than all other nodes) to emphasize its importance and guide algorithms to start reconstruction from this point. (EGF is currently not in the data). We can assign it a score of 10; chosen empirically.
EGFR is in the current data. Looking at the pathway, we can see that EGFR directly interacts with EGF in the pathway.
All other downstream proteins detected in the data can also treated as targets.
All proteins in the data can be considered active since they correspond to proteins that are active under the given biological condition.
Input node data transformed into a SPRAS-standardized format:
NODE_ID prize source target active
AAGAB_HUMAN 0.906857382 True True
... more nodes
EGF_HUMAN 10 True True True
EGFR_HUMAN 6.787874699 True True
... more nodes
1.8 Finding an Interactome to use
To connect our proteins, we need a background interactome. For this dataset, we merge two protein-protein interaction (PPI) interactomes, prioritizing directed edges when both sources include the same interaction:
iRefIndex v13 (159,095 undirected interactions)
PhosphoSitePlus (4,080 directed kinase-substrate interactions)
The final network has 15,677 proteins and 157,984 edges (~4k of them are directed), and covers 653 of our 702 prize proteins. The proteins identifiers in the interactome are converted to use UniProt Entry Names.
Interactome data put into a SPRAS-standardized format:
TACC1_HUMAN RUXG_HUMAN 0.736771 U
TACC1_HUMAN KAT2A_HUMAN 0.292198 U
TACC1_HUMAN CKAP5_HUMAN 0.724783 U
TACC1_HUMAN YETS4_HUMAN 0.542597 U
TACC1_HUMAN LSM7_HUMAN 0.714823 U
AURKC_HUMAN TACC1_HUMAN 0.553333 D
TACC1_HUMAN AURKA_HUMAN 0.401165 U
TACC1_HUMAN KDM1A_HUMAN 0.367850 U
TACC1_HUMAN MEMO1_HUMAN 0.367850 U
TACC1_HUMAN HD_HUMAN 0.367850 U
... more edges
Note
Many databases provide interactomes. One example is STRING, which contains known protein-protein interactions across different species. For a broader overview of available interactomes, see Koh et al. (2025). Users can also construct their own interactomes from experimental or curated data.
1.9 This SPRAS-standardized data is already saved into SPRAS
spras/
├── .snakemake/
│ └── log/
│ └── ... snakemake log files ...
├── config/
│ └── ...
├── inputs/
│ ├── phosphosite-irefindex13.0-uniprot.txt # pre-defined in SPRAS already, used by the intermediate.yaml file
│ └── tps-egfr-prizes.txt # pre-defined in SPRAS already, used by the intermediate.yaml file
├── outputs/
│ └── basic/
│ └── ... output files ...
The data used in this part of the tutorial can be found in the supplementary materials under data supplement 2 and supplement 3 [4].
Step 2: Running multiple algorithms
We can begin running multiple pathway reconstruction algorithms.
For this part of the tutorial, we’ll use a pre-defined configuration
file that includes additional algorithms and post-analysis steps
available in SPRAS. Download it here: Intermediate Config
File
Save the file into the config/ folder of your SPRAS installation.
After adding this file, your directory structure will look like this (ignoring the rest of the folders):
spras/
├── .snakemake/
│ └── log/
│ └── ... snakemake log files ...
├── config/
│ ├── basic.yaml
│ ├── intermediate.yaml
│ └── ... other configs ...
├── inputs/
│ ├── phosphosite-irefindex13.0-uniprot.txt # pre-defined in SPRAS already, used by the intermediate.yaml file
│ ├── tps-egfr-prizes.txt # pre-defined in SPRAS already, used by the intermediate.yaml file
│ └── ... other input data ...
├── outputs/
│ └── basic/
│ └── ... output files ...
2.1 Algorithms in SPRAS
SPRAS supports a wide range of algorithms, each designed around different biological assumptions and optimization strategies (See Pathway Reconstruction Methods for SPRAS’s list of integrated algorithms.)
Wrapped algorithms
Each pathway reconstruction algorithm within SPRAS has been wrapped for SPRAS, meaning it has been prepared for the SPRAS framework.
For an algorithm-specific wrapper, the wrapper includes a module that will create and format the input files required by the algorithm using the SPRAS-standardized input data.
Each algorithm has an associated Docker image located on DockerHub that contains all necessary
software dependencies needed to run it. For an algorithm-specific
wrapper, it contains a module that will call each image to launch a
container for a specified parameter combination, set of prepared
algorithm-specific inputs and an output filename (raw-pathway.txt).
With each of the raw-pathway.txt files, an algorithm-specific
wrapper includes a module that will convert the algorithm-specific
format into a standardized SPRAS output format.
2.3 Running SPRAS with multiple algorithms
In the intermediate.yaml configuration file, it is set up to have
SPRAS run multiple algorithms with multiple parameter settings on a
single dataset.
algorithms:
- name: "pathlinker"
include: true
runs:
run1:
k: [1, 10, 100, 1000]
- name: omicsintegrator2
include: true
runs:
run1:
b: [4, 10]
g: [0, 3]
w: [0.25, 6]
- name: mincostflow
include: true
runs:
run1:
capacity: [15, 30]
flow: [80, 15]
- name: "strwr"
include: true
runs:
run1:
alpha: 0.85
threshold: [100, 200]
- name: "rwr"
include: true
runs:
run1:
alpha: 0.85
threshold: [100, 200]
Note
The full suite of algorithms is described in Pathway Reconstruction Methods. This part of the tutorial uses only a subset.
From the root directory, run the command below from the command line:
snakemake --cores 4 --configfile config/intermediate.yaml
What happens when you run this command
SPRAS will run “slower” when using the intermediate.yaml
configuration.
Similar automated steps from the previous tutorial runs behind the
scenes for intermediate.yaml. However, this configuration now runs
multiple algorithms with different parameter combinations, which takes
longer to complete. By increasing the number of cores to 4, it allows
Snakemake to parallelize the work locally, speeding up execution when
possible. (See Using SPRAS for more information on
SPRAS’s parallelization.)
Snakemake starts the workflow
Snakemake reads the options set in the intermediate.yaml
configuration file and determines which datasets, algorithms, and
parameter combinations need to run. It also checks if any post-analysis
steps were requested.
Creating algorithm-specific inputs
For each algorithm marked as include: true in the configuration,
SPRAS generates input files tailored to that algorithm.
In this case, every algorithm is enabled, so SPRAS formats the input files required for each algorithm.
Organizing results with parameter hashes
Each <dataset>-<algorithm>-params-<hash> combination gets its own folder
created in output/intermediate/.
A matching log file in
logs/parameters-<algorithm>-params-<hash>.yaml records the exact
parameter values used.
Running the algorithm
SPRAS pulls each algorithm’s Docker image from DockerHub if it isn’t already downloaded locally
SPRAS executes each algorithm by launching multiple Docker contatiners using the algorithm-specific Docker image (once for each parameter configuration), sending the prepared input files and specific parameter settings needed for execution.
Each algorithm runs independently within its Docker container and
generates an output file named raw-pathway.txt, which contains the
reconstructed subnetwork in the algorithm-specific format.
SPRAS then saves these files to the corresponding folder.
Standardizing the results
SPRAS parses each of the raw output into a standardized SPRAS format
(pathway.txt) and SPRAS saves this file in its corresponding folder.
Logging the Snakemake run
Snakemake creates a dated log in .snakemake/log/ This log shows what
jobs ran and any errors that occurred during the SPRAS run.
What your directory structure should like after this run:
spras/
├── .snakemake/
│ └── log/
│ └── ... snakemake log files ...
├── config/
│ └── basic.yaml
├── inputs/
│ ├── phosphosite-irefindex13.0-uniprot.txt
│ └── tps-egfr-prizes.txt
├── outputs/
│ └── intermediate/
│ ├── dataset-egfr-merged.pickle
│ ├── egfr-mincostflow-params-42UBTQI/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-B4P4LUU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-KTZPGLQ/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-MY6UCHG/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-44PJEHW/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-4NC62EL/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-4VRLTK5/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-52OUGT2/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-KEVHYWP/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-RUGOWNI/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-RVH2YKU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-WW2ILRO/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-7S4SLU6/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-D4TUKMX/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-TFORORH/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-VQL7BDZ/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-rwr-params-34NN6EK/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-rwr-params-GGZCZBU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-strwr-params-34NN6EK/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-strwr-params-GGZCZBU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── logs/
│ │ ├── datasets-egfr.yaml
│ │ ├── parameters-mincostflow-params-42UBTQI.yaml
│ │ ├── parameters-mincostflow-params-B4P4LUU.yaml
│ │ ├── parameters-mincostflow-params-KTZPGLQ.yaml
│ │ ├── parameters-mincostflow-params-MY6UCHG.yaml
│ │ ├── parameters-omicsintegrator2-params-44PJEHW.yaml
│ │ ├── parameters-omicsintegrator2-params-4NC62EL.yaml
│ │ ├── parameters-omicsintegrator2-params-4VRLTK5.yaml
│ │ ├── parameters-omicsintegrator2-params-52OUGT2.yaml
│ │ ├── parameters-omicsintegrator2-params-KEVHYWP.yaml
│ │ ├── parameters-omicsintegrator2-params-RUGOWNI.yaml
│ │ ├── parameters-omicsintegrator2-params-RVH2YKU.yaml
│ │ ├── parameters-omicsintegrator2-params-WW2ILRO.yaml
│ │ ├── parameters-pathlinker-params-7S4SLU6.yaml
│ │ ├── parameters-pathlinker-params-D4TUKMX.yaml
│ │ ├── parameters-pathlinker-params-TFORORH.yaml
│ │ ├── parameters-pathlinker-params-VQL7BDZ.yaml
│ │ ├── parameters-rwr-params-34NN6EK.yaml
│ │ ├── parameters-rwr-params-GGZCZBU.yaml
│ │ ├── parameters-strwr-params-34NN6EK.yaml
│ │ └── parameters-strwr-params-GGZCZBU.yaml
│ └── prepared/
│ ├── egfr-mincostflow-inputs/
│ │ ├── edges.txt
│ │ ├── sources.txt
│ │ └── targets.txt
│ ├── egfr-omicsintegrator2-inputs/
│ │ ├── edges.txt
│ │ └── prizes.txt
│ ├── egfr-pathlinker-inputs/
│ │ ├── network.txt
│ │ └── nodetypes.txt
│ ├── egfr-rwr-inputs/
│ │ ├── network.txt
│ │ └── nodes.txt
│ └── egfr-strwr-inputs/
│ ├── network.txt
│ ├── sources.txt
│ └── targets.txt
2.4 Reviewing the pathway.txt files
After running the intermediate configuration file, the
output/intermediate/ directory will contain many more subfolders and
files.
Again, each pathway.txt file contains the standardized reconstructed
subnetworks and can be used at face value, or for further post analysis.
Locate the files
Navigate to the output directory output/intermediate/. Inside, you
will find subfolders corresponding to each
<dataset>-<algorithm>-params-<hash> combination.
Open a
pathway.txtfile
Each file lists the network edges that were reconstructed for that specific run. The format includes columns for the two interacting nodes, the rank, and the edge direction.
For example, the file egfr-mincostflow-params-42UBTQI/pathway.txt
contains the following reconstructed subnetwork:
Node1 Node2 Rank Direction
CBL_HUMAN EGFR_HUMAN 1 U
EGFR_HUMAN EGF_HUMAN 1 U
EMD_HUMAN LMNA_HUMAN 1 U
FYN_HUMAN KS6A3_HUMAN 1 U
EGF_HUMAN HDAC6_HUMAN 1 U
HDAC6_HUMAN HS90A_HUMAN 1 U
KS6A3_HUMAN SRC_HUMAN 1 U
EGF_HUMAN LMNA_HUMAN 1 U
MYH9_HUMAN S10A4_HUMAN 1 U
EGF_HUMAN S10A4_HUMAN 1 U
EMD_HUMAN SRC_HUMAN 1 U
And the file egfr-omicsintegrator1-params-YYFFQV4/pathway.txt
contains the following reconstructed subnetwork:
Node1 Node2 Rank Direction
CBLB_HUMAN EGFR_HUMAN 1 U
CBL_HUMAN CD2AP_HUMAN 1 U
CBL_HUMAN CRKL_HUMAN 1 U
CBL_HUMAN EGFR_HUMAN 1 U
CBL_HUMAN PLCG1_HUMAN 1 U
CDK1_HUMAN NPM_HUMAN 1 D
CHD4_HUMAN HDAC1_HUMAN 1 U
CHIP_HUMAN HS90A_HUMAN 1 U
CHIP_HUMAN P53_HUMAN 1 U
DNMT1_HUMAN HDAC1_HUMAN 1 U
EGFR_HUMAN EGF_HUMAN 1 U
EGFR_HUMAN GRB2_HUMAN 1 U
EIF3B_HUMAN EIF3G_HUMAN 1 U
FAK1_HUMAN PAXI_HUMAN 1 U
GAB1_HUMAN PTN11_HUMAN 1 U
GRB2_HUMAN KHDR1_HUMAN 1 U
GRB2_HUMAN PTN11_HUMAN 1 U
GRB2_HUMAN SHC1_HUMAN 1 U
HDAC1_HUMAN HDAC2_HUMAN 1 U
HDAC1_HUMAN P53_HUMAN 1 U
HDAC1_HUMAN RB_HUMAN 1 U
HDAC1_HUMAN SIN3A_HUMAN 1 U
HGS_HUMAN STAM2_HUMAN 1 U
HS90A_HUMAN STIP1_HUMAN 1 U
HS90A_HUMAN TEBP_HUMAN 1 U
KHDR1_HUMAN LCK_HUMAN 1 U
KS6A1_HUMAN MK01_HUMAN 1 U
MK01_HUMAN ABI1_HUMAN 1 D
MK01_HUMAN ERF_HUMAN 1 D
MRE11_HUMAN RAD50_HUMAN 1 U
P53_HUMAN TP53B_HUMAN 1 U
Step 3: Use ML post-analysis
Rather than inspecting each output on its own, users may want to understand how the outputs from multiple algorithms and parameter combinations relate to one another when run on the same dataset. SPRAS includes machine learning (ML) post-analysis methods for this: hierarchical agglomerative clustering, principal component analysis, Jaccard similarity, and ensembling.
Note
Each ML method operates on a dataset-specific binary edge-by-subnetwork matrix. Rows represent edges in the union of all reconstructed pathways, and columns represent output subnetworks. An entry indicates whether a given edge appears in a given subnetwork (1 if present, 0 if absent).
3.1 Adding ML post-analysis to the intermediate configuration
To enable the ML analysis, update the analysis section in your configuration file by setting ML to true. Your analysis section in the configuration file should look like this:
analysis:
ml:
include: true
... (other parameters preset)
ml will perform unsupervised analyses such as principal component
analysis, hierarchical agglomerative clustering, ensembling, and jaccard
similarity comparisons of the pathways.
Note
The ml section includes configurable parameters that adjust the
behavior of these analyses. For the available options, see
config.yaml in the config/ folder.
With these updates, SPRAS will run the full ML analyses across all outputs for a given dataset.
After saving the changes in the configuration file, rerun with:
snakemake --cores 4 --configfile config/intermediate.yaml
What happens when you run this command
Reusing cached results
Snakemake reads the options set in intermediate.yaml and checks for
any requested post-analysis steps. It reuses cached results; here the
pathway.txt files generated from the previously executed algorithms
on the egfr dataset are reused.
Running the ML analysis
SPRAS aggregates all the reconstructed subnetworks produced across the
specified algorithms for a given dataset. SPRAS then performs machine
learning analyses on each these groups and saves the results in a
<dataset>-ml/ (egfr-ml/ in this case) folder.
What your directory structure should like after this run:
spras/
├── .snakemake/
│ └── log/
│ └── ... snakemake log files ...
├── config/
│ └── basic.yaml
├── inputs/
│ ├── phosphosite-irefindex13.0-uniprot.txt
│ └── tps-egfr-prizes.txt
├── outputs/
│ └── intermediate/
│ ├── dataset-egfr-merged.pickle
│ ├── egfr-mincostflow-params-42UBTQI/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-B4P4LUU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-KTZPGLQ/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-MY6UCHG/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-ml/
│ │ ├── ensemble-pathway.txt
│ │ ├── hac-clusters-horizontal.txt
│ │ ├── hac-clusters-vertical.txt
│ │ ├── hac-horizontal.png
│ │ ├── hac-vertical.png
│ │ ├── jaccard-heatmap.png
│ │ ├── jaccard-matrix.txt
│ │ ├── pca-coordinates.txt
│ │ ├── pca-variance.txt
│ │ └── pca.png
│ ├── egfr-omicsintegrator2-params-44PJEHW/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-4NC62EL/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-4VRLTK5/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-52OUGT2/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-KEVHYWP/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-RUGOWNI/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-RVH2YKU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-WW2ILRO/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-7S4SLU6/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-D4TUKMX/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-TFORORH/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-VQL7BDZ/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-rwr-params-34NN6EK/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-rwr-params-GGZCZBU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-strwr-params-34NN6EK/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-strwr-params-GGZCZBU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── logs/
│ │ ├── datasets-egfr.yaml
│ │ ├── parameters-mincostflow-params-42UBTQI.yaml
│ │ ├── parameters-mincostflow-params-B4P4LUU.yaml
│ │ ├── parameters-mincostflow-params-KTZPGLQ.yaml
│ │ ├── parameters-mincostflow-params-MY6UCHG.yaml
│ │ ├── parameters-omicsintegrator2-params-44PJEHW.yaml
│ │ ├── parameters-omicsintegrator2-params-4NC62EL.yaml
│ │ ├── parameters-omicsintegrator2-params-4VRLTK5.yaml
│ │ ├── parameters-omicsintegrator2-params-52OUGT2.yaml
│ │ ├── parameters-omicsintegrator2-params-KEVHYWP.yaml
│ │ ├── parameters-omicsintegrator2-params-RUGOWNI.yaml
│ │ ├── parameters-omicsintegrator2-params-RVH2YKU.yaml
│ │ ├── parameters-omicsintegrator2-params-WW2ILRO.yaml
│ │ ├── parameters-pathlinker-params-7S4SLU6.yaml
│ │ ├── parameters-pathlinker-params-D4TUKMX.yaml
│ │ ├── parameters-pathlinker-params-TFORORH.yaml
│ │ ├── parameters-pathlinker-params-VQL7BDZ.yaml
│ │ ├── parameters-rwr-params-34NN6EK.yaml
│ │ ├── parameters-rwr-params-GGZCZBU.yaml
│ │ ├── parameters-strwr-params-34NN6EK.yaml
│ │ └── parameters-strwr-params-GGZCZBU.yaml
│ └── prepared/
│ ├── egfr-mincostflow-inputs/
│ │ ├── edges.txt
│ │ ├── sources.txt
│ │ └── targets.txt
│ ├── egfr-omicsintegrator2-inputs/
│ │ ├── edges.txt
│ │ └── prizes.txt
│ ├── egfr-pathlinker-inputs/
│ │ ├── network.txt
│ │ └── nodetypes.txt
│ ├── egfr-rwr-inputs/
│ │ ├── network.txt
│ │ └── nodes.txt
│ └── egfr-strwr-inputs/
│ ├── network.txt
│ ├── sources.txt
│ └── targets.txt
Step 3.2: Reviewing the ML outputs
Ensembles
Open the ensemble file
In your file explorer, go to
output/intermediate/egfr-ml/ensemble-pathway.txt and open it
locally.
After running multiple algorithms or parameter settings on the same dataset, SPRAS can ensemble the resulting pathways to identify consistent, high-frequency interactions. SPRAS calculates the edge frequency by calculating the proportion of times each edge appears across the outputs.
Node1 Node2 Frequency Direction
EGF_HUMAN EGFR_HUMAN 0.3 D
EGF_HUMAN S10A4_HUMAN 0.25 D
S10A4_HUMAN MYH9_HUMAN 0.2 D
K7PPA8_HUMAN MDM2_HUMAN 0.15 D
MDM2_HUMAN P53_HUMAN 0.15 D
S10A4_HUMAN K7PPA8_HUMAN 0.15 D
K7PPA8_HUMAN SIR1_HUMAN 0.15 D
MDM2_HUMAN MDM4_HUMAN 0.15 D
MDM4_HUMAN P53_HUMAN 0.15 D
CD2A2_HUMAN CDK4_HUMAN 0.15 D
CDK4_HUMAN RB_HUMAN 0.15 D
MDM2_HUMAN CD2A2_HUMAN 0.15 D
EP300_HUMAN P53_HUMAN 0.35 D
K7PPA8_HUMAN EP300_HUMAN 0.15 D
...
High frequency edges indicate interactions consistently recovered by multiple algorithms. Low frequency edges may reflect noise or algorithm-specific connections. Edges that occur across many outputs are less likely to be algorithm-specific artifacts, so ensembling lets you filter for interactions supported by multiple algorithms or parameter settings.
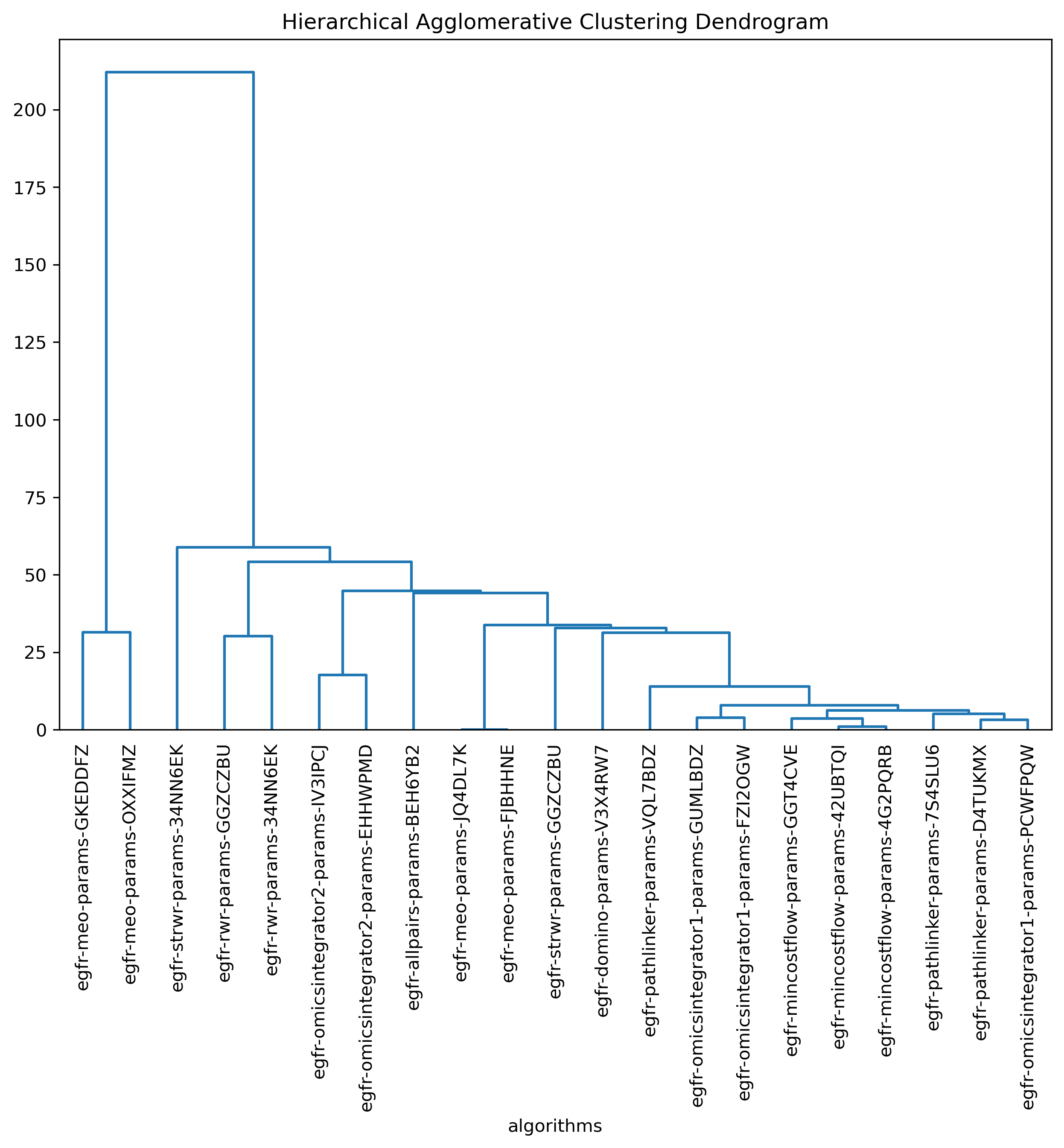
Hierarchical agglomerative clustering
Open the hierarchical agglomerative clustering image(s)
In your file explorer, go to
output/intermediate/egfr-ml/hac-horizontal.png and/or
output/intermediate/egfr-ml/hac-vertical.png and open it locally.
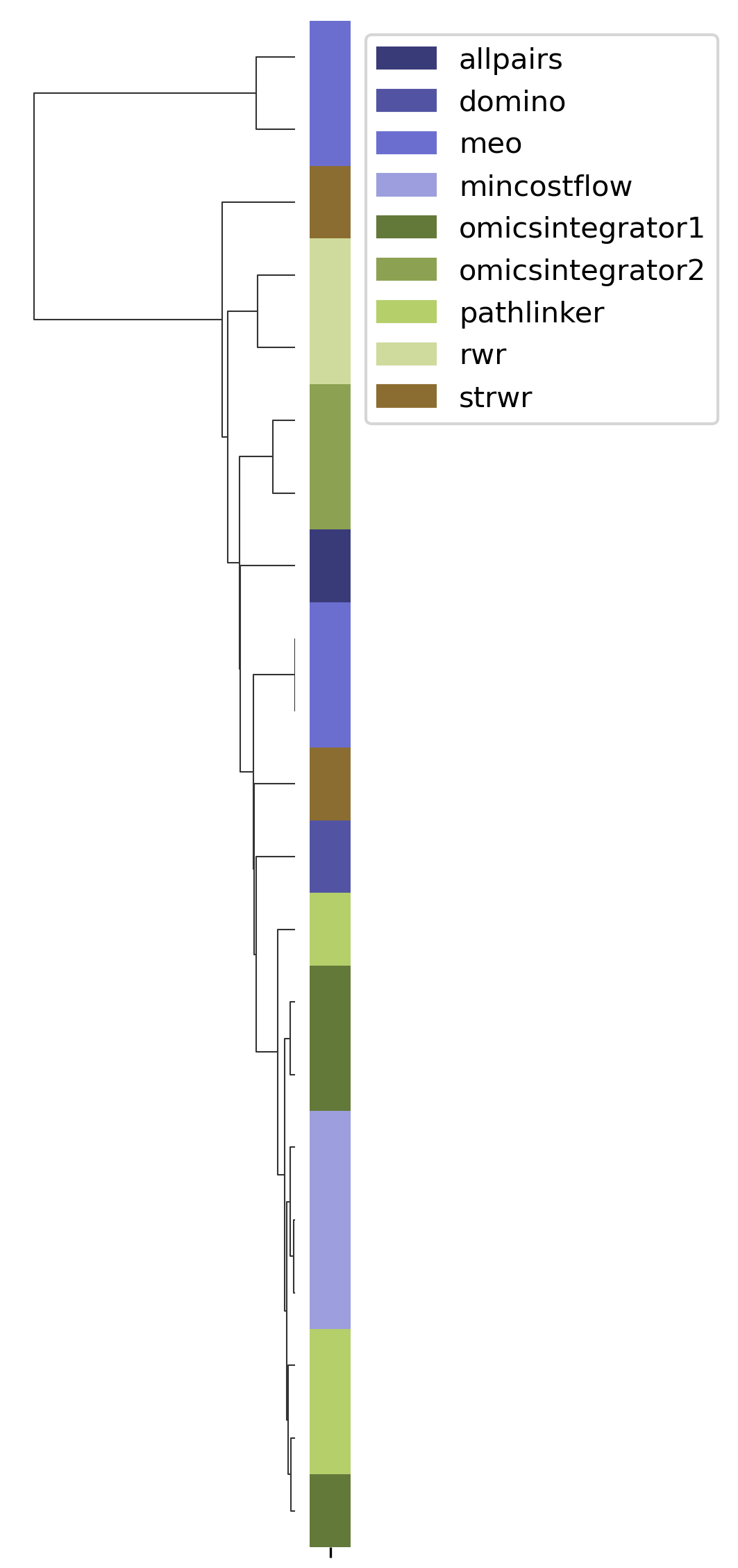
SPRAS includes hierarchical agglomerative clustering to group similar pathways outputs based on shared edges. This helps identify clusters of algorithms that produce comparable subnetworks and highlights distinct reconstruction behaviors.
In the plots below, each branch represents a cluster of related pathways, and shorter distances between branches indicate greater similarity. Tight clusters group algorithms and parameter settings that produce comparable pathway structures, while isolated branches flag outputs that differ from the rest.
Principal component analysis
Open the PCA image
In your file explorer, go to output/intermediate/egfr-ml/pca.png and
open it locally.
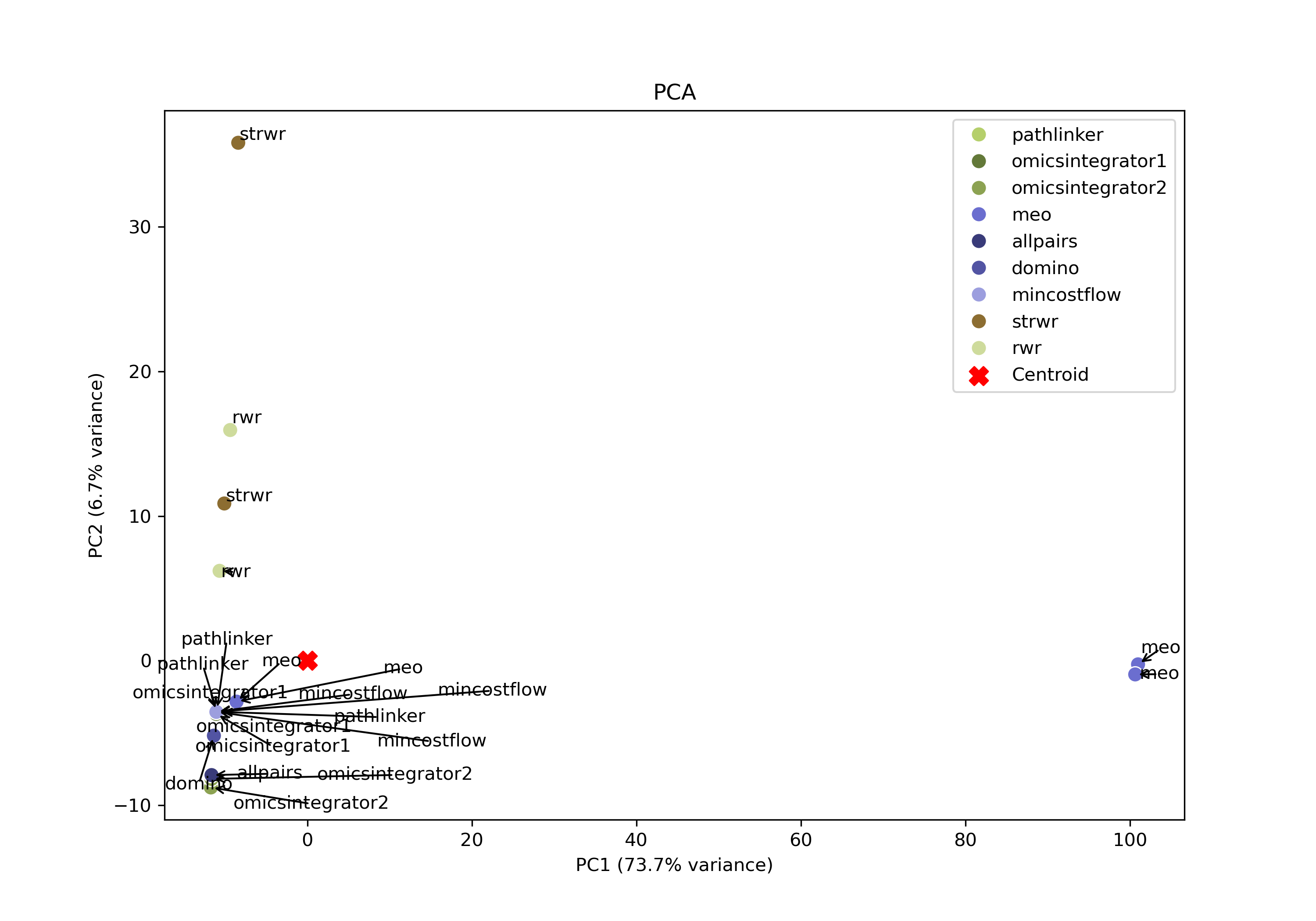
SPRAS also includes principal component analysis (PCA) to visualize variation across pathway outputs. Each point represents a pathway, placed based on its overall network structure. Pathways that cluster together in PCA space are more similar, while those farther apart differ in their reconstructed subnetworks. PCA may help identify patterns such as clusters of similar algorithms outputs, parameter sensitivities, and/or outlier outputs in a lower lower-dimensional space.
Jaccard similarity
Open the jaccard heatmap image
In your file explorer, go to
output/intermediate/egfr-ml/jaccard-heatmap.png and open it locally.
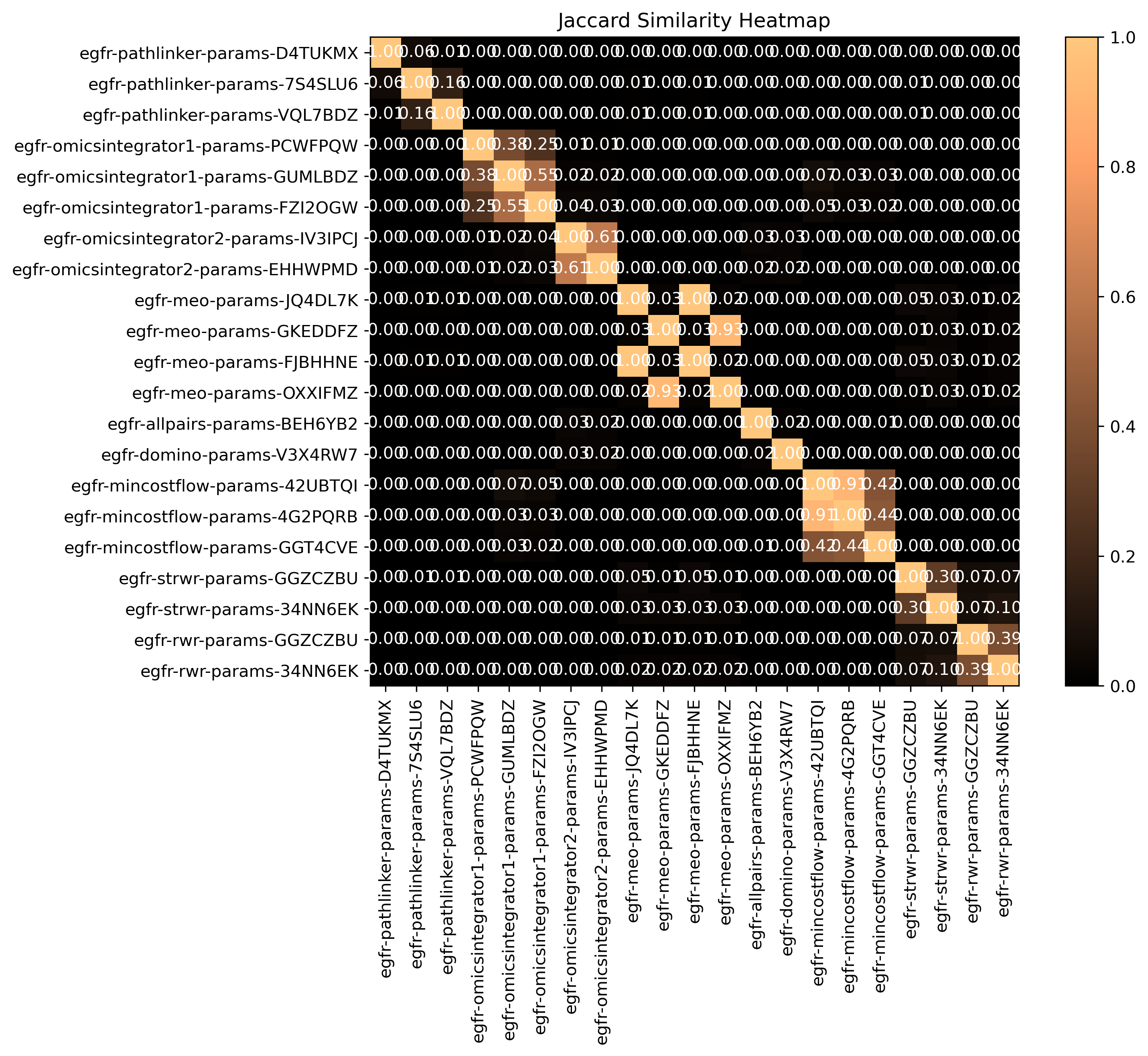
SPRAS computes pairwise jaccard similarity between pathway outputs to measure how much overlap exists between their reconstructed subnetworks. The heatmap visualizes how similar the output pathways are between algorithms and their parameter settings. Higher similarity values indicate that pathways share many of the same edges, while lower values suggest distinct reconstructions.
Step 4: Use Evaluation post-analysis
In some cases, users may have a gold standard file that allows them to evaluate the quality of the reconstructed subnetworks generated by pathway reconstruction algorithms.
However, gold standards may not exist for certain types of experimental data where validated ground truth interactions or molecules are unavailable or incomplete. For example, in emerging research areas or poorly characterized biological systems, interactions may not yet be experimentally verified or fully known, making it difficult to define a reliable reference network for evaluation.
Note
A gold standard captures interactions that are already known, but pathway reconstruction is also a tool for discovery. An algorithm that scores well against a gold standard may do so by recovering established interactions while missing novel ones.
4.1 Adding evaluation post-analysis to the intermediate configuration
To enable evaluation, update the analysis section of your configuration
file. In the evaluation section, set include and
aggregate_per_algorithm to true. Also, in the ml section,
set kde, r`emove_empty_pathways, and aggregate_per_algorithm
to true. Your analysis section in the configuration file should look
like this:
analysis:
summary:
include: true
ml:
include: true
aggregate_per_algorithm: true
kde: true
remove_empty_pathways: true
evaluation:
include: true
aggregate_per_algorithm: true
Setting aggregate_per_algorithm to true will additionally group
post-analysis and evaluations by algorithm per dataset. Without this,
outputs from all algorithm per dataset are aggregated together for
post-analysis rather than broken out per algorithm.
Within ml, remove_empty_pathways excludes pathways with no nodes
or edges from the PCA post analysis. The kde creates a kernel
density estimate over the PCA plots.
summary is enabled because evaluation uses summary statistics to
break ties between pathways for some of the parameter selection methods
(more details further into the tutorial).
We need to delete the existing egfr-ml/ folder before rerunning
SPRAS so that Snakemake regenerates the ML outputs with the new
customized ML settings. Run this command from the root directory:
rm -rf output/intermediate/egfr-ml/
Note
Snakemake skips steps whose output files already exist, so changes to ML configuration parameters will not trigger a rerun unless the existing ML outputs are removed first.
Automatic re-execution on config changes is a known limitation and is being addressed in ongoing SPRAS development.
The intermediate configuration also includes a gold standard for the EGFR dataset, which is already set up in SPRAS and does not require any setup:
gold_standards:
-
label: gs_egfr
node_files: ["gs-egfr.txt"]
data_dir: "input"
dataset_labels: ["egfr"]
Note
The gold standard for this dataset consists of nodes only, following the original study. The gold standard nodes are drawn from eight EGFR-related reference pathways [4].
A limitation of this gold standard is its incomplete coverage of EGF signaling pathways. Across the eight reference pathways, typically 5% or fewer of the input nodes appear in any single pathway, and 85% are absent from all eight. This reflects the general incompleteness of curated pathway databases relative to measured signaling responses, rather than a flaw specific to this dataset [4].
With these updates, SPRAS will run the evaluations across all outputs for a given dataset.
After saving the changes in the configuration file, rerun with:
snakemake --cores 4 --configfile config/intermediate.yaml
What happens when you run this command
Reusing cached results
Snakemake reads the options set in intermediate.yaml and checks for
any requested post-analysis steps. It reuses cached results; here the
pathway.txt files generated from the previously executed algorithms
on the egfr dataset are reused.
Running the ML analysis
SPRAS aggregates all the reconstructed subnetworks produced across the
specified algorithms for a given dataset. SPRAS then performs machine
learning analyses on each these groups and saves the results in the
<dataset>-ml/ (egfr-ml/) folder. It is also going to be running
the ML per algorithm for a given dataset. This groups the ML post
analysis by algorithm per dataset and produces algorithm specific ML
outputs.
Running the summary analysis
SPRAS aggregates the pathway.txt files from all selected parameter
combinations into a single summary table, saved as
egfr-pathway-summary.txt. This is used if any tiebreakers occur
during PCA-based parameter selection.
Running the evaluation
For each dataset listed in a gold standard’s dataset_labels, SPRAS
compares the reconstructed subnetworks against that gold standard using
the parameter selection methods enabled in the configuration.
The evaluation runs at two levels: once across all algorithms combined,
and once per algorithm. The per-algorithm evaluation depends on
per-algorithm ML outputs, which is why aggregate_per_algorithm was
set to true in the ml section above. This produces both
all-algorithm evaluation files and algorithm-specific evaluation files
for each dataset-goldstandard pair.
What your directory structure should like after this run:
spras/
├── .snakemake/
│ └── log/
│ └── ... snakemake log files ...
├── config/
│ └── basic.yaml
├── inputs/
│ ├── phosphosite-irefindex13.0-uniprot.txt
│ └── tps-egfr-prizes.txt
├── outputs/
│ └── intermediate/
│ ├── dataset-egfr-merged.pickle
│ ├── egfr-gs_egfr-eval/
│ │ ├── pr-curve-ensemble-nodes-per-algorithm-nodes.png
│ │ ├── pr-curve-ensemble-nodes-per-algorithm-nodes.txt
│ │ ├── pr-curve-ensemble-nodes.png
│ │ ├── pr-curve-ensemble-nodes.txt
│ │ ├── pr-pca-chosen-pathway-nodes.png
│ │ ├── pr-pca-chosen-pathway-nodes.txt
│ │ ├── pr-pca-chosen-pathway-per-algorithm-nodes.png
│ │ ├── pr-pca-chosen-pathway-per-algorithm-nodes.txt
│ │ ├── pr-per-pathway-for-mincostflow-nodes.png
│ │ ├── pr-per-pathway-for-mincostflow-nodes.txt
│ │ ├── pr-per-pathway-for-omicsintegrator2-nodes.png
│ │ ├── pr-per-pathway-for-omicsintegrator2-nodes.txt
│ │ ├── pr-per-pathway-for-pathlinker-nodes.png
│ │ ├── pr-per-pathway-for-pathlinker-nodes.txt
│ │ ├── pr-per-pathway-for-rwr-nodes.png
│ │ ├── pr-per-pathway-for-rwr-nodes.txt
│ │ ├── pr-per-pathway-for-strwr-nodes.png
│ │ ├── pr-per-pathway-for-strwr-nodes.txt
│ │ ├── pr-per-pathway-nodes.png
│ │ └── pr-per-pathway-nodes.txt
│ ├── egfr-mincostflow-params-42UBTQI/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-B4P4LUU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-KTZPGLQ/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-mincostflow-params-MY6UCHG/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-ml/
│ │ ├── ensemble-pathway.txt
│ │ ├── hac-clusters-horizontal.txt
│ │ ├── hac-clusters-vertical.txt
│ │ ├── hac-horizontal.png
│ │ ├── hac-vertical.png
│ │ ├── jaccard-heatmap.png
│ │ ├── jaccard-matrix.txt
│ │ ├── mincostflow-ensemble-pathway.txt
│ │ ├── mincostflow-hac-clusters-horizontal.txt
│ │ ├── mincostflow-hac-clusters-vertical.txt
│ │ ├── mincostflow-hac-horizontal.png
│ │ ├── mincostflow-hac-vertical.png
│ │ ├── mincostflow-jaccard-heatmap.png
│ │ ├── mincostflow-jaccard-matrix.txt
│ │ ├── mincostflow-pca-coordinates.txt
│ │ ├── mincostflow-pca-variance.txt
│ │ ├── mincostflow-pca.png
│ │ ├── omicsintegrator2-ensemble-pathway.txt
│ │ ├── omicsintegrator2-hac-clusters-horizontal.txt
│ │ ├── omicsintegrator2-hac-clusters-vertical.txt
│ │ ├── omicsintegrator2-hac-horizontal.png
│ │ ├── omicsintegrator2-hac-vertical.png
│ │ ├── omicsintegrator2-jaccard-heatmap.png
│ │ ├── omicsintegrator2-jaccard-matrix.txt
│ │ ├── omicsintegrator2-pca-coordinates.txt
│ │ ├── omicsintegrator2-pca-variance.txt
│ │ ├── omicsintegrator2-pca.png
│ │ ├── pathlinker-ensemble-pathway.txt
│ │ ├── pathlinker-hac-clusters-horizontal.txt
│ │ ├── pathlinker-hac-clusters-vertical.txt
│ │ ├── pathlinker-hac-horizontal.png
│ │ ├── pathlinker-hac-vertical.png
│ │ ├── pathlinker-jaccard-heatmap.png
│ │ ├── pathlinker-jaccard-matrix.txt
│ │ ├── pathlinker-pca-coordinates.txt
│ │ ├── pathlinker-pca-variance.txt
│ │ ├── pathlinker-pca.png
│ │ ├── pca-coordinates.txt
│ │ ├── pca-variance.txt
│ │ ├── pca.png
│ │ ├── rwr-ensemble-pathway.txt
│ │ ├── rwr-hac-clusters-horizontal.txt
│ │ ├── rwr-hac-clusters-vertical.txt
│ │ ├── rwr-hac-horizontal.png
│ │ ├── rwr-hac-vertical.png
│ │ ├── rwr-jaccard-heatmap.png
│ │ ├── rwr-jaccard-matrix.txt
│ │ ├── rwr-pca-coordinates.txt
│ │ ├── rwr-pca-variance.txt
│ │ ├── rwr-pca.png
│ │ ├── strwr-ensemble-pathway.txt
│ │ ├── strwr-hac-clusters-horizontal.txt
│ │ ├── strwr-hac-clusters-vertical.txt
│ │ ├── strwr-hac-horizontal.png
│ │ ├── strwr-hac-vertical.png
│ │ ├── strwr-jaccard-heatmap.png
│ │ ├── strwr-jaccard-matrix.txt
│ │ ├── strwr-pca-coordinates.txt
│ │ ├── strwr-pca-variance.txt
│ │ └── strwr-pca.png
│ ├── egfr-omicsintegrator2-params-44PJEHW/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-4NC62EL/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-4VRLTK5/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-52OUGT2/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-KEVHYWP/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-RUGOWNI/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-RVH2YKU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-omicsintegrator2-params-WW2ILRO/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-7S4SLU6/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-D4TUKMX/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-TFORORH/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathlinker-params-VQL7BDZ/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-pathway-summary.txt
│ ├── egfr-rwr-params-34NN6EK/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-rwr-params-GGZCZBU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-strwr-params-34NN6EK/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── egfr-strwr-params-GGZCZBU/
│ │ ├── pathway.txt
│ │ └── raw-pathway.txt
│ ├── gs-gs_egfr-merged.pickle
│ ├── logs/
│ │ ├── datasets-egfr.yaml
│ │ ├── parameters-mincostflow-params-42UBTQI.yaml
│ │ ├── parameters-mincostflow-params-B4P4LUU.yaml
│ │ ├── parameters-mincostflow-params-KTZPGLQ.yaml
│ │ ├── parameters-mincostflow-params-MY6UCHG.yaml
│ │ ├── parameters-omicsintegrator2-params-44PJEHW.yaml
│ │ ├── parameters-omicsintegrator2-params-4NC62EL.yaml
│ │ ├── parameters-omicsintegrator2-params-4VRLTK5.yaml
│ │ ├── parameters-omicsintegrator2-params-52OUGT2.yaml
│ │ ├── parameters-omicsintegrator2-params-KEVHYWP.yaml
│ │ ├── parameters-omicsintegrator2-params-RUGOWNI.yaml
│ │ ├── parameters-omicsintegrator2-params-RVH2YKU.yaml
│ │ ├── parameters-omicsintegrator2-params-WW2ILRO.yaml
│ │ ├── parameters-pathlinker-params-7S4SLU6.yaml
│ │ ├── parameters-pathlinker-params-D4TUKMX.yaml
│ │ ├── parameters-pathlinker-params-TFORORH.yaml
│ │ ├── parameters-pathlinker-params-VQL7BDZ.yaml
│ │ ├── parameters-rwr-params-34NN6EK.yaml
│ │ ├── parameters-rwr-params-GGZCZBU.yaml
│ │ ├── parameters-strwr-params-34NN6EK.yaml
│ │ └── parameters-strwr-params-GGZCZBU.yaml
│ └── prepared/
│ ├── egfr-mincostflow-inputs/
│ │ ├── edges.txt
│ │ ├── sources.txt
│ │ └── targets.txt
│ ├── egfr-omicsintegrator2-inputs/
│ │ ├── edges.txt
│ │ └── prizes.txt
│ ├── egfr-pathlinker-inputs/
│ │ ├── network.txt
│ │ └── nodetypes.txt
│ ├── egfr-rwr-inputs/
│ │ ├── network.txt
│ │ └── nodes.txt
│ └── egfr-strwr-inputs/
│ ├── network.txt
│ ├── sources.txt
│ └── targets.txt
4.2 What is parameter selection?
Parameter selection refers to the process of determining which parameter combinations should be used for evaluation on a gold standard dataset. Each parameter selection method has its own corresponding evaluation procedure.
Note
There is no single principled way to decide which outputs to evaluate, so SPRAS provides several parameter selection strategies instead of committing to one. Some strategies pick a single representative output for each algorithm, while others evaluate across the full set of parameter combinations.
Parameter selection also guards against overtuning. Algorithms differ in how many parameters they expose and how much they can be tuned to get a better answer, so comparing them on a representative output rather than on the full sweep puts them on some fairer footing.
Selecting a representative output also measures how an algorithm typically behaves rather than its best run, which is a better basis for judging an algorithm in practice, where the ideal parameters for a new dataset are not known in advance.
Parameter selection is handled in the evaluation code, which supports
multiple parameter selection strategies. A user can enable evaluation
(by setting evaluation include: true) and it will run all of the
parameter selection code.
Note
Some parameter selection features are still under development and will be added in future SPRAS releases.
PCA-based parameter selection
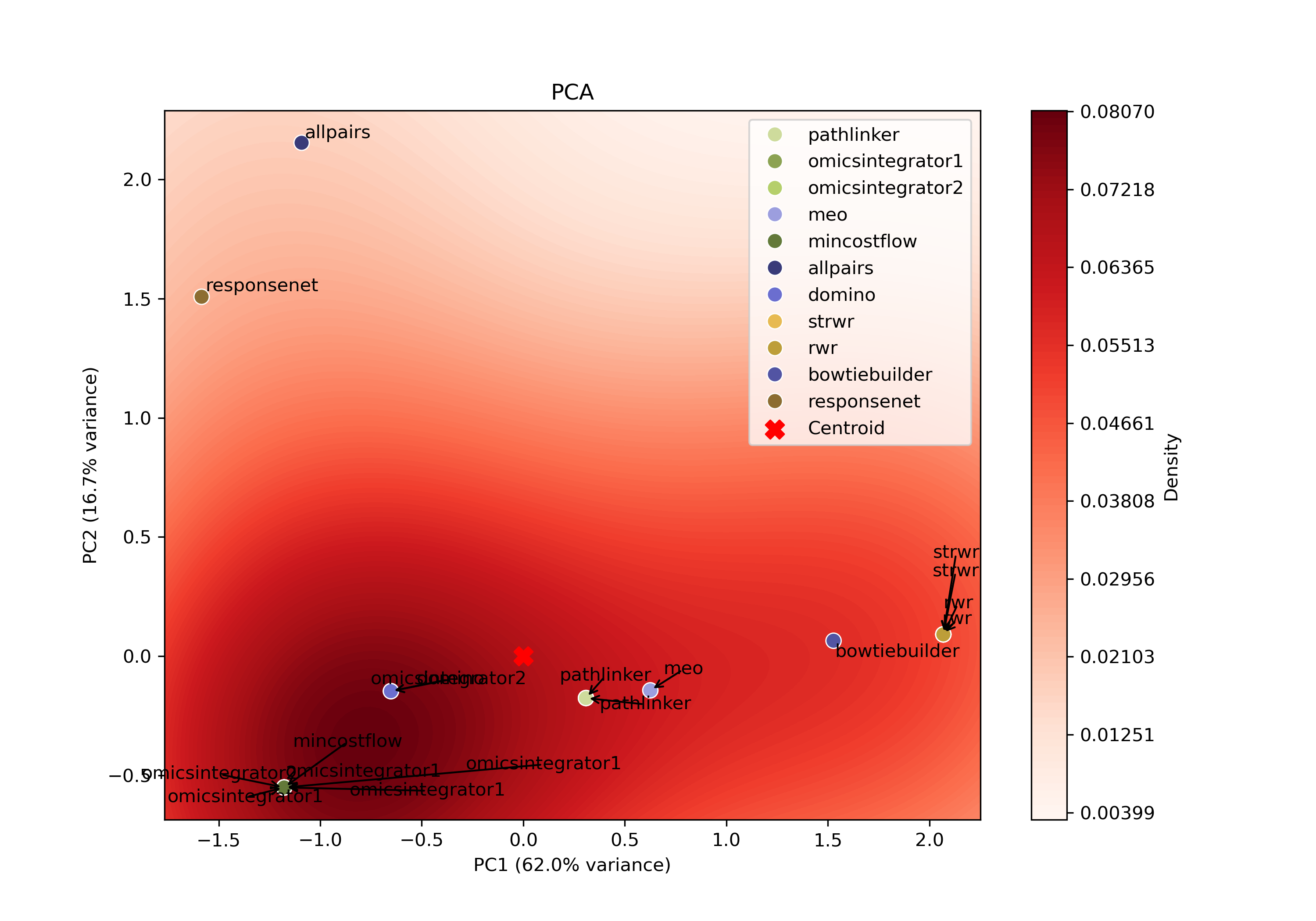
The PCA-based approach identifies a representative parameter setting for each pathway reconstruction algorithm on a given dataset. It selects the single parameter combination that best captures the central trend of an algorithm’s reconstruction behavior.
For each algorithm, all reconstructed subnetworks are projected into an algorithm-specific 2D PCA space based on the set of edges produced by the respective parameter combinations for that algorithm. This projection summarizes how the algorithm’s outputs vary across different parameter combinations, allowing patterns in the outputs to be visualized in a lower-dimensional space.
Within each PCA space, a kernel density estimate (KDE) is computed over the projected points to identify regions of high density. The output closest to the highest KDE peak is selected as the most representative parameter setting, as it corresponds to the region where the algorithm most consistently produces similar subnetworks.
If two or more pathways are equally close to the highest peak of the KDE, SPRAS resolves the tie by:
Choosing the smallest pathway (fewest nodes and edges).
If a tie remains, choosing the first pathway alphabetically by name.
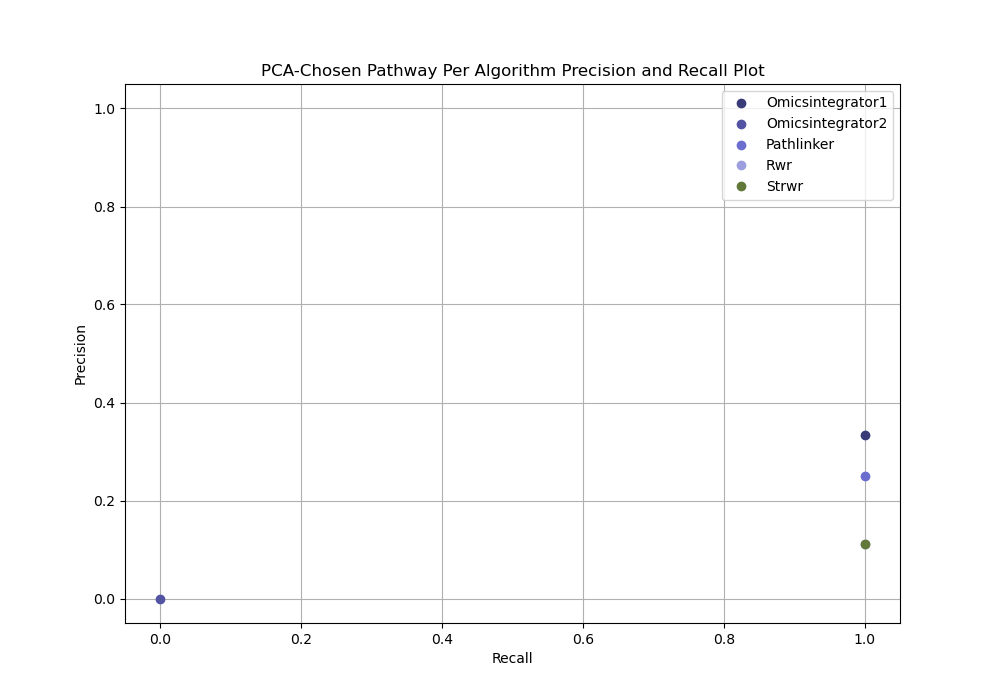
The chosen output subnetwork is then compared to the gold standard, and its precision and recall are measured.
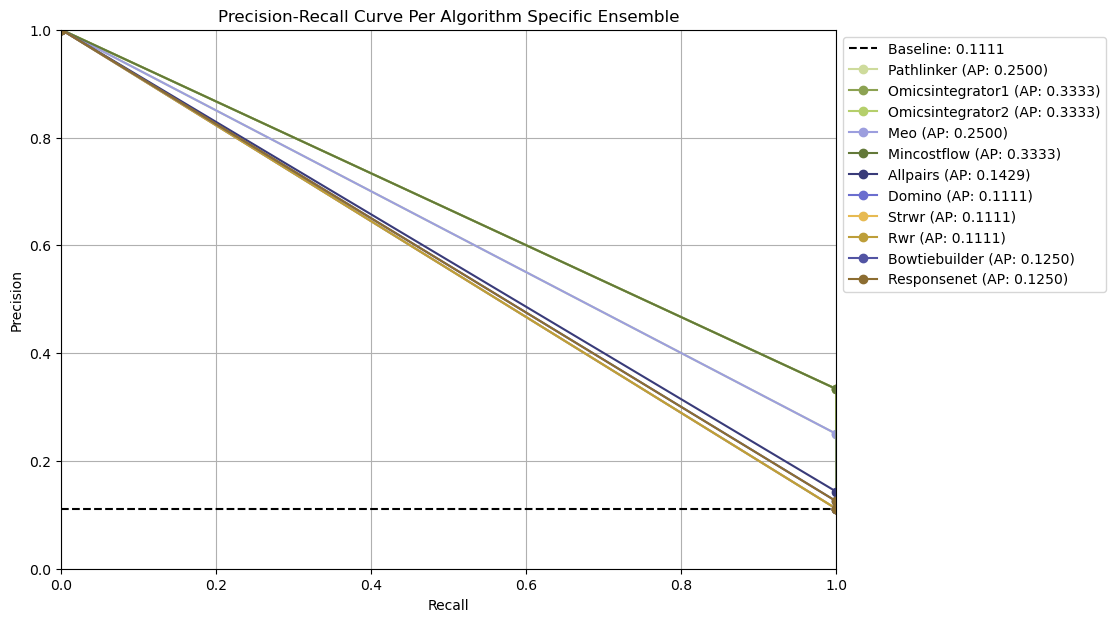
Ensemble network-based parameter selection
The ensemble-based approach combines results from all parameter settings for each pathway reconstruction algorithm on a given dataset. Instead of focusing on a single “best” parameter combination, it summarizes the algorithm’s overall reconstruction behavior across parameters.
All reconstructed subnetworks are merged into algorithm-specific ensemble networks, where each edge weight reflects how frequently that interaction appears across the outputs. Edges that occur more often are assigned higher weights, highlighting interactions that are most consistently recovered by the algorithm.
These consensus networks help identify the core patterns and overall stability of an algorithm’s output’s without needing to choose a single parameter setting (no clear optimal parameter combination could exists).
For each algorithm-specific ensemble network, SPRAS generates a precision-recall curve by treating edge frequencies as thresholds and evaluating each ensemble network against the dataset’s associated gold standard.
All Plausible Parameters (No parameter selection)
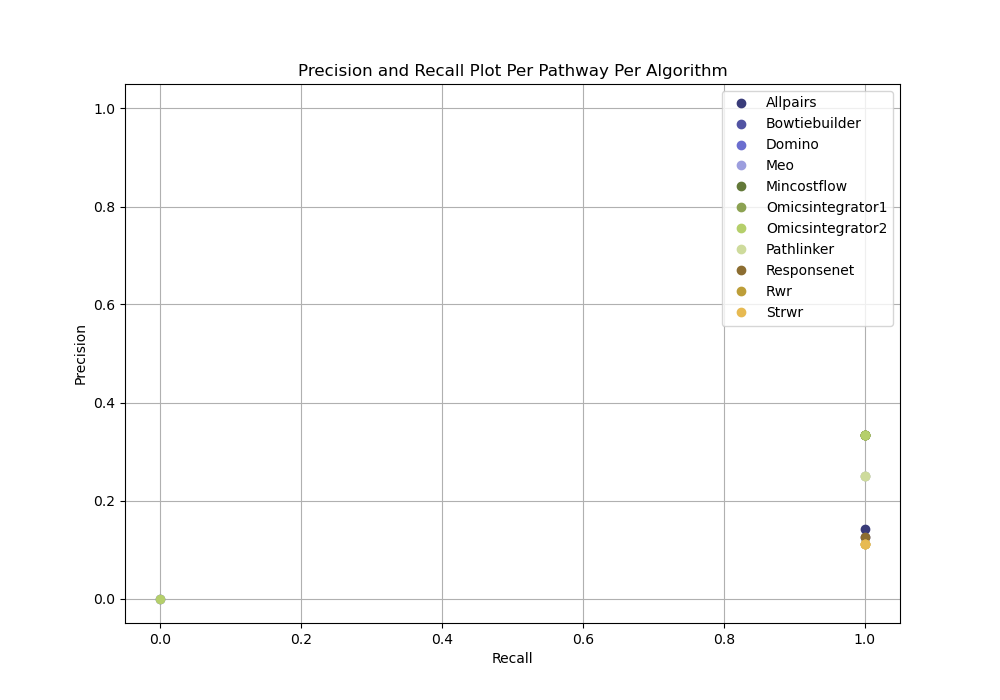
The all plausible parameters approach evaluates all parameter combinations without selecting a representative subset or ensembling. This method provides an holistic view of algorithm performance by evaluating every output. For each algorithm and dataset, we compute precision and recall for every subnetwork. This allows us to measure reconstruction performance across the full range of parameter settings and observe each algorithm’s full range of capabilities.
4.4 Reviewing the evalaution outputs
PCA-based parameter selection
Open the PCA chosen parameter selection evaluation
In your file explorer, go to
output/intermediate/egfr-gs_egfr-eval/pr-per-pathway-nodes.png and
open it locally.
PCA-based parameter selection computes a precision and recall for a single reconstructed network selected using PCA from all reconstructed networks for an algorithm for given dataset.
Ensemble network-based parameter selection
Open the Ensemble-based parameter selection evalaution
In your file explorer, go to
output/intermediate/egfr-gs_egfr-eval/pr-curve-ensemble-nodes-per-algorithm-nodes.png
and open it locally.
Ensemble-based parameter selection generates precision-recall curves by thresholding on the frequency of edges across an ensemble of reconstructed networks for an algorithm for given dataset.
All Plausible Parameters (No parameter selection)
Open the all plausible parameters (no parameter selection) evalaution
In your file explorer, go to
output/intermediate/egfr-gs_egfr-eval/pr-per-pathway-nodes.png and
open it locally.
For each pathway, evaluation can be run independently of any parameter selection method to directly inspect precision and recall for each reconstructed network from a given dataset.